When the Data Doesn't Hold Up: Inside Replimune's Second FDA Rejection
A small biotech, an oncolytic virus, and a four-year regulatory story that's now ending in a media campaign instead of a randomized trial.

The FDA has been beset by controversy in the Trump administration over the perception, reinforced repeatedly in outlets like the Wall Street Journal, that it is acting in a chaotic, unpredictable manner when it comes to regulating the pharmaceutical industry. As evidence, the story of the biotech company Replimune is proffered — what’s been called a breakthrough drug for metastatic melanoma that received its second rejection by the Makary-led FDA. Yesterday, a CNBC reporter pressed Makary hard on the basis of the rejection. A frustrated Makary touched on some of the reasons but repeatedly referred to the FDA letters that served as the basis for the rejections.
I’m not an oncologist, but I read the FDA CRL letters, and boy, this story has been completely mangled by the media. At the very least, they appear unable or unwilling to do anything but recycle Replimune’s talking points. Given that the Wall Street Journal is essentially calling for Makary to be fired over this, here’s my summary of the affair, detailing the very strong FDA case against approval of Replimune’s drug.
Replimune is a small biotech firm whose lead asset is vusolimogene oderparepvec (RP1), an oncolytic HSV-1 virus injected directly into tumors with the idea of provoking both a local kill and a systemic anti-tumor immune response. The proposed indication is a real unmet need population: advanced cutaneous melanoma in patients who have already progressed on anti-PD1 therapy.
Replimune wants to give their drug in combination with another drug called nivolumab. That combination is where the trouble starts.
The Combination Problem
Nivolumab (Opdivo, Bristol Myers Squibb) is a PD-1 blocking antibody approved in melanoma since 2014, used both as monotherapy and in combination with ipilimumab, and one of the foundational drugs in the disease. The wrinkle is that the patients enrolled in Replimune’s trial had already progressed on a PD-1 inhibitor — some on nivolumab, some on pembrolizumab, some on combination checkpoint blockade, with prior therapy given in either the adjuvant or metastatic setting. So the trial was layering RP1 onto a drug class these patients had already failed.
That sounds like it should make nivolumab’s contribution easy to dismiss — if they already failed PD-1, nivolumab can’t be doing anything, right?
Except continuing or rechallenging anti-PD1 therapy after progression is not zero-activity. There’s a real signal in the literature that some patients regain sensitivity, especially when combined with another agent that may reverse resistance. The activity isn’t well-quantified, which is precisely the problem: you can’t subtract out an unknown number. Without a control arm, every response in RPL-001-16 has two plausible explanations and no way to choose between them.
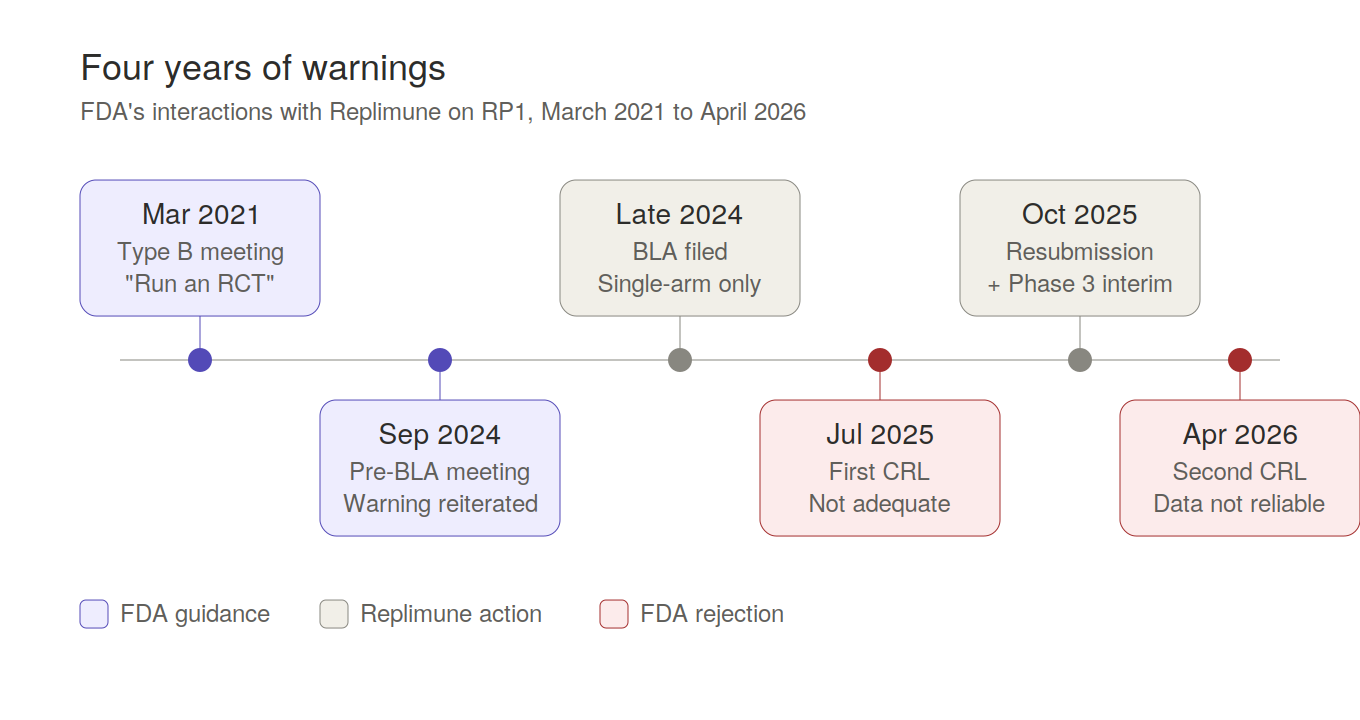
Four Years of Warnings
The pivotal data came from RPL-001-16, a single-arm Phase 2 of 140 patients, no concurrent control, with objective response rate as the primary endpoint. Replimune filed a BLA on this and received their first Complete Response Letter in July 2025. To understand why, you have to go back to March 2021, because FDA had been telegraphing this outcome for four years.
At a Type B meeting in March 2021, FDA told Replimune in writing that a single-arm study would not allow identification of each component’s contribution to response rate, and explicitly recommended a randomized controlled trial. They flagged that a 30-40% ORR target might not translate to meaningful clinical benefit, that baseline patient heterogeneity would confound interpretation, and that interpreting responses in injected lesions was inherently problematic. They stated directly they would not recommend submitting a BLA based on a single-arm study.
FDA reiterated all of this at the pre-BLA meeting in September 2024.
Replimune filed the BLA on the single-arm Phase 2 anyway, apparently betting that the unmet need narrative plus a numerically attractive ORR would carry approval. FDA didn’t formally object to the filing — that’s the regulatory flexibility piece — but flexibility about accepting a filing is not the same as flexibility about substantial evidence of effectiveness. The July 2025 CRL said exactly what you’d expect: not adequate and well-controlled, contribution of RP1 versus nivolumab can’t be isolated, go run a real trial.
Replimune resubmitted in October 2025 with exploratory reanalyses and an early interim look at their Phase 3, and that resubmission is what got rejected again last month — with the second CRL going considerably further than the first.
The Phase 3 “Rescue”
The October 2025 resubmission was a two-part rescue effort: exploratory reanalyses of the original RPL-001-16 data, plus an early unplanned interim look at their ongoing Phase 3 (RP1-104) showing 22 patients on RP1+nivo versus 18 on physician’s choice.
That’s 10% of the planned 400-patient enrollment, with response assessed by investigators rather than blinded review, no duration of response data, and a progression-free survival analysis that was never pre-specified and had no type 1 error control. FDA’s response on this piece was essentially that an unplanned 10% interim from an ongoing trial isn’t evidence of effectiveness, it’s a status update.
But the more damaging part of the second CRL is what FDA found when they reanalyzed the Phase 2 data itself.
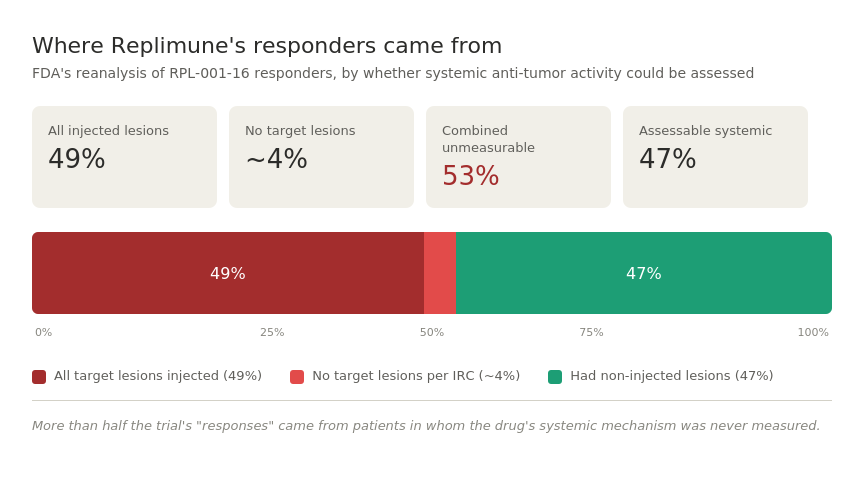
The 53% Problem
A different review team was assigned to the resubmission to avoid bias from the first round, and they pulled the individual patient data apart. The headline finding:
49% of patients counted as responders had every single one of their target lesions injected with RP1, and another two responders had no target lesions at all per independent review. That means 53% of responders had no non-injected lesions to assess systemic anti-tumor activity — the entire premise of an oncolytic virus working through the immune system rather than just locally.
This requires understanding how RP1 is supposed to work.
An oncolytic virus like RP1 is injected directly into a tumor — you literally stick a needle into a visible or accessible lesion (skin lesion, lymph node, etc.) and inject the virus. Two things are then supposed to happen:
Local effect: the virus replicates inside tumor cells, lyses them, and the tumor at the injection site shrinks. This is mechanically obvious — you injected a tumor-killing agent directly into a tumor. Of course it shrinks.
Systemic effect: the lysed tumor cells release tumor antigens, which prime the immune system to recognize and attack tumor cells elsewhere in the body — including tumors that were never injected. This is the abscopal effect, and it’s the entire scientific rationale for developing oncolytic viruses as a class. If all you wanted was local tumor destruction you could use surgery, radiation, cryoablation, or any number of cheaper, simpler interventions.
The whole reason FDA would approve a systemic-acting oncolytic immunotherapy is the second mechanism. The first one isn’t novel and isn’t clinically meaningful for patients with metastatic disease, where the threat is distant tumors you can’t reach with a needle.
How Response Is Measured
In oncology trials, response is measured using RECIST criteria. At baseline, the radiologist identifies a small number of “target lesions” (typically up to five) that will be measured at every subsequent scan. These are the lesions whose changes in size determine whether the patient has a partial response, complete response, stable disease, or progression. Non-target lesions and new lesions also factor in but are assessed more qualitatively.
The point of target lesions is they’re your measurement substrate. Whatever happens to them is what gets called the response.
So what did the FDA find when they independently reviewed the primary patient-level data?
49% of responders had every single target lesion injected with RP1. Meaning: the lesions being measured to determine whether the drug “worked” were the same lesions that had a virus needle physically stuck into them. Of course those lesions shrank. The drug was deposited directly into them.
Weirdly enough, two more responders had no target lesions at all per independent review at baseline. So these were people counted as responders that shouldn’t have been based on independent review.
Combined, almost 53% of responders — more than half of the “wins” in the trial — came from patients in whom systemic anti-tumor activity was not measured, either because every measured lesion was injected, or because there were no measured lesions at all.
If you wanted to design a trial that was specifically engineered to not test the systemic effect of an oncolytic virus, this is roughly what you’d do: make sure the lesions you’re measuring are the same ones you’re injecting, and count those local responses toward your primary endpoint.
The right way to design this trial — and I imagine the FDA almost certainly told them this — is to require each patient to have at least one non-injected target lesion at baseline. That non-injected lesion is your real readout. If it shrinks, you’ve got evidence of systemic immune activation (the abscopal effect, the actual drug mechanism). If it doesn’t shrink while the injected lesion does, all you’ve shown is that injecting a virus into a tumor kills the tumor at the injection site, which nobody doubted.
When you exclude these patients from the primary endpoint analysis, the response rate drops markedly from what Replimune reported. FDA’s framing here is very polite: “the systemic anti-tumor activity of RP1 is not well characterized.”
Translation: a meaningful share of the reported responses may just be tumors shrinking because you injected them with a virus, which is a local effect any oncolytic agent should produce and which doesn’t require a randomized trial or a BLA to demonstrate.
It Gets Worse
FDA found patients who were re-injected with RP1 after developing new lesions or having existing lesions enlarge, but before the independent review committee had called progression. Re-treating before progression is officially declared can extend the apparent duration of response past the actual progression event and count post-progression responses as if they were initial responses.
They also found excisional biopsies and surgical procedures performed on target lesions during the treatment period, sometimes immediately preceding the response assessment that determined a partial or complete response. Removing tumor tissue and then measuring tumor size shortly afterward is not a measure of drug activity.
Add in histopathologic assessments that were not centrally reviewed but were used to override radiologic response calls, plus deviations from standard RECIST 1.1 methodology, and FDA’s conclusion writes itself: “the reported response rate and duration of response from RPL-001-16 were not reliably assessed, could be artifactually increased, and may not be reproducible.”
What This Actually Means
The first CRL said the trial design can’t answer the question. The second CRL says even setting design aside, the numbers themselves don’t hold up.
The takeaway isn’t really about FDA flexibility, or unmet need, or whether oncolytic viruses are a promising modality. The company was told in 2021 to run a randomized trial. Told again in 2024. Ran the single-arm trial anyway. The FDA politely chose to review the data that was submitted to see if there was something amazing going on, and on close inspection of the primary patient-level data the responses the company says happened don’t even cleanly attribute to systemic drug activity.
The Media Campaign
And yet the major media story in The Wall Street Journal, Squawk Box, and across the Replimune investor community — who need their stock positions rescued — is that this is FDA malfeasance.
Completely absurd.
If the drug is that amazing, raise some more money to run an RCT and prove it. Except no one with half a brain who looks at this timeline would put any trust in a company that has made all these harebrained decisions. So the company has pulled the only emergency parachute possible: flood journalists who cover this beat with stories about FDA in chaos, in the hope of getting Commissioner Makary fired and sending a message to the next FDA commissioner — approve Replimune or else.
Absolutely nuts.
If this does come to pass, just end the FDA already, and let people try whatever they want. There’s apparently a huge number of biotech investors who care tremendously about rare disease patients and will put up a lot of money to fund these therapies.
Anish Koka is a cardiologist in Philadelphia. He writes on medicine and healthcare policy and hosts a weekly podcast : The Doctor’s Lounge
Great piece.
This company is an abomination and an embarrassment. As are all the “journalists” carrying water for it.
Thank you for another outstanding piece following your thorough summary of the Vinay Prasad travesty. You might think that “science journalists” dedicated to the FDA beat might have at least a cursory understanding of trial design. What a pathetic display of ignorance. Thanks for making your voice heard.